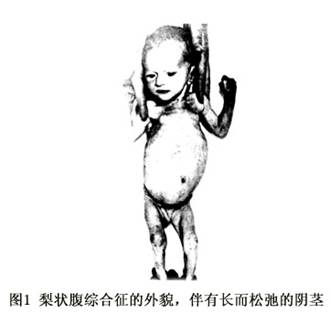
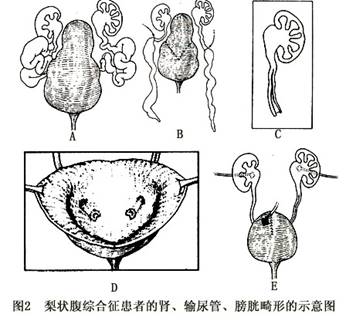
流行病学
流行病学
流行病学:3.5万~5万活产男婴中有1例。1970年前约半数是死后确诊的,其中20%是死产或在新生儿期死亡,另30%于2岁内死亡,多死于
尿路感染败血症或肾功能衰竭或二者兼有。
患先天性腹壁肌肉发育不良的婴儿可出生于正常妊娠的母体,但更多见于羊水量少以及伴发其他复杂情况尤以肺及骨骼方面。因做产前超声检查日益增多,故本症可于宫内诊断。
近年本症预后有所改进,治疗意见尚不统一,有些学者主张积极手术修复尿路,另有学者主张仅做有限而必要的手术。
女性先天性腹壁肌肉发育不良文献上报告甚少。严格来说,完全的综合征只发生于男性,但有报告3%发生于女性,可能实际的发病率更高些。
病因
病因:本症的原因不明,有些作者提出腹壁缺陷及腹腔内睾丸都是继发于早期胎儿发育期的尿路扩张。后尿道梗阻可引起尿路扩张并可解释本症见于男性,因膀胱扩张阻止睾丸下降而腹壁缺陷继发于尿路扩张或胎儿
腹水。但其他严重的后尿道梗阻如
后尿道瓣膜症并不发生相似的腹壁缺陷或相似的上尿路迂曲。此外,
隐睾的高发率也未见于其他梗阻性尿路病变。
有些作者提出前列腺发育不良及胎儿
腹水是引起本症的主要因素,胎儿
腹水导致腹壁缺陷。梨状腹综合征患者却罕见
腹水,推测
腹水是一过性的,妊娠期胎儿
腹水吸收。另一风靡一时的说法:原始中胚层的误差造成腹壁缺陷及尿路畸形,因为这二者都起源于轴旁间质中胚层及中胚层板。有些作者则提出本症是因复杂的染色体突变所致。本症的基因基础仍有争论,有作者认为是单基因缺陷,亦有认为是不同基因产生相同的结果。实际上,不少证据支持性别连锁遗传的说法,已有一些报告在同胞间发生梨状腹综合征。所有报告孪生儿并发本症者不一致。如梨状腹综合征患者有慢性肾功能不全须做肾移植时,供肾须选择正常单合子孪生者。
多数本症患者的核型是正常的。极少数报告
染色体异常,包括本症的同胞间有嵌合体及合并Turner综合征。
发病机制
发病机制:
1.肾脏 肾脏可以正常,但肾发育异常和
肾积水是本综合征的典型表现。肾发育异常可见于约一半的患者,而且具多样性与不对称性。尤其合并
尿道狭窄、巨尿道或肛门闭锁时肾发育异常更为明显。多数患者有不同程度的与腹壁缺陷程度无关的
肾积水,由于仅少数患者合并肾盂、输尿管连接部梗阻,而且肾实质厚度,肾功能损害程度与积水程度并不平行,推测本症
肾积水与典型的尿路梗阻所致者机制不同,肾实质损害的最大危险可能来自反复发生的感染而非梗阻。
2.输尿管 输尿管表现为扭曲、节段性扩张或狭窄,一般近端输尿管显示正常,远端输尿管病变为重。显微镜下观察,病变组织平滑肌细胞数量减少,代之以纤维结缔组织,神经丛数目也减少,而且超微病理发现粗、细肌丝数目均减少。上述异常的不利影响在于输尿管蠕动功能障碍,导致尿液淤积易于诱发感染,而且用输尿管下段做重建术时易导致手术失败。
3.膀胱和脐尿管 输尿管口位于正常位置的后外侧,
膀胱输尿管反流发生率可达85%(Carret,1978)。膀胱壁肥厚但膀胱肌肉并无增生,无膀胱小梁形成;常合并脐尿管憩室而使膀胱增大,膀胱颈宽大,当有尿道闭锁时,可有脐尿管开放,但有些患儿在脐尿管开放的同时并无尿道梗阻。
尿流动力学检查显示逼尿肌顺应性良好,灌注末压正常、逼尿肌压力降低、残余尿、膀胱容量增大等。有些患者随年龄增长可改善排尿状况,有些则逐渐加重(图2)。
4.前列腺和附属性腺 前列腺发育异常,影像学表现为前列腺尿道扩张,渐至膜部变细,类似
后尿道瓣膜的病理改变。但实际上后尿道闭锁、狭窄和瓣膜在存活的新生儿中罕见。在一组胎儿、新生儿尸检中发现合并本病者,部分病例精囊退化或缺如、输精管闭锁、附睾与睾丸连接处较纤细。
5.尿道 绝大部分梨状腹综合征患者前尿道和阴茎正常,偶有
先天性巨尿道。海绵体发育不良和海绵体缺如亦见报道。
6.睾丸 双侧
隐睾是本症特点,多数患儿
隐睾位于腹腔内平骨盆缘近骶髂关节水平,覆在扩张输尿管之前。扩张尿路的阻碍、引带缺如和睾丸自身缺陷为其可能的原因,患者阴茎勃起和性高潮正常,但多合并逆行射精,尚未见男性生育的报道。
7.其他异常 腹壁缺陷:本症的特点是患者腹直肌上部及腹外斜肌发育,腹肌在腹壁下内侧缺如。尽管外形相似,但患者腹壁缺陷可有较大差别,常呈不对称片状分布。有些研究者认为其肌肉存在但发育异常,另有人认为其肌肉缺如。Nunn研究认为患者肌神经前根是完整的。就手术而言,尽管患者腹壁缺乏层次以利缝合,愈合过程却多较满意,极少发生腹壁裂开。由于下腹壁缺乏支持,不能进行有效的咳嗽,患者容易患呼吸道感染。
肺脏:患先天性腹壁肌肉发育不良的新生儿肺部状况常威胁其生存。肺发育异常合并
羊水过少时尤其严峻。其他肺部并发症尚有肺叶不张和肺炎等。
心脏:本症患者房、室间隔缺损,法洛四联症的发生率远高于正常人群,可高达10%。
胃肠道:许多患者有肠旋转不良、肛门闭锁而且多合并于尿道闭锁及肾发育异常。腹壁与巨结肠症亦有报道。患者常受
便秘困扰,也可发生继发性巨结肠症。
骨骼:本综合征患者最常见膝外侧皮肤形成小凹,其次为畸形足。臀部发育异常合并
先天性髋关节脱位及脊柱畸形各占约5%。
临床表现
临床表现:患儿于胎儿30周时即有典型的表现:扩张的输尿管,大膀胱以及松弛的腹壁,产前B超可发现,但难与原发性
膀胱输尿管反流或
后尿道瓣膜相鉴别。
新生儿期临床表现差别很大,轻者虽有尿路病变,但肾实质结构及功能良好,尿淤滞较轻,也可有严重而广泛的尿路扩张,肾发育异常,但限于单侧,严重的有肾发育异常,肺发育不全,可有完全尿道梗阻及脐尿管开放,有Potter面容,血清肌酐量正常,但出生后不断增高,可发生死产或在新生儿期死于肺发育不全。临床常将患儿按症状严重程度分为三大类:
Ⅱ型:典型梨状腹外表,全尿路扩张,但尚未危及生命。可有轻度或
单侧肾发育不良,可有脓毒症或进行性氮质血症。
Ⅲ型:外型不典型或不完全,尿道症状不严重,肾功能尚稳定。
女性患者的主要表现在于腹壁、膀胱及上尿路的缺陷,其尿道一般是正常的。很多女性患者,综合征不完全而上尿路正常。Lubinsky等(1980)曾报告1例Turner综合征(核型XO)患者并发本症。
不完全先天性腹壁肌肉发育不良在男性患儿可见有典型的腹壁缺陷,但尿路却正常,这种情况很罕见。但相反的情况并不少见。有些病人(假性梨状腹综合征)有正常或相对正常的腹壁,却有很多或全部不太严重的尿路异常,包括肾形态异常、扩张迂曲的输尿管、大容量膀胱以及典型膀胱颈及前列腺部尿道改变。
治疗
治疗:理论上讲,超声诊断胎儿患本症时,在宫内就应开始治疗。但这样处理并未证明肾及肺的功能发生改变。目前婴儿梨状腹综合征的治疗始于新生儿期的最初评估。
1.非手术治疗 如果排除梗阻和膀胱输尿管反流,且肾功能正常,即便存在明显的尿路扩张,亦可采用非手术治疗。可应用预防性抗生素,但要监测肾功能、有无感染、尿路扩张的进展及有无梗阻出现等。
2.手术治疗 梨状腹综合征尿路损害的外科治疗的目的在于纠正膀胱输尿管、膀胱尿道功能失常,如膀胱输尿管反流、上尿路梗阻、严重尿液淤滞等。如存在梗阻和感染,宜早期手术,否则可稍晚进行。所有患者均需行睾丸下降固定术。是否采用腹壁重建要依据腹壁缺陷程度而定。
(1)暂时性尿流改道:若感染难以控制或肾功能恶化,需行暂时性尿流改道。膀胱造瘘术为首选,当存在肾盂输尿管连接部、输尿管膀胱壁段狭窄或持续感染时可选用高位引流术,如有可能,尽量不用近段输尿管造瘘以免影响后续的重建术。
(2)输尿管重建术:尿路重建术应用在本症患者尚有争议,有些人未行手术而生存良好,而接受不成功手术者则适得其反。由于尿液淤滞及感染可以损害肾功能,相当多的医生仍倾向于尿路重建。尿路重建术成功与否的关键在于最大限度的利用正常或相对正常的近段输尿管。要细致操作,按重建原则裁剪和再植输尿管。
(3)缩小膀胱容量:膀胱成形术目的在于增加逼尿肌收缩力。可采用切除脐尿管憩室、膀胱折叠术等。
(4)尿道内切开术:尿道内切开术适用于尿道梗阻及膀胱尿道协同失调所致的残余尿量增多、膀胱输尿管反流或输尿管扩张的患者。可应用Otis尿道内切开术或以内腔镜进行。
(5)巨大尿道的修复:前尿道显著扩张罕见,多位于阴茎部尿道,尿道口及阴茎头部正常,须将其恢复正常口径。
(6)隐睾手术:尽管本症患者生育能力值得怀疑,但研究显示睾丸内确存在干细胞,而且睾丸的内分泌功能对患者进入青春期必不可少。大部分患者需行睾丸下降固定术,可采用3种方法:
①早期经腹腔睾丸下降固定术:适用于新生儿及6个月以内婴儿,多作为尿路重建术的同期手术。如不作尿路重建术,可待3~4个月后上尿路状况及心肺功能稳定后再行手术。
②Fowler-Stephens术:本术式已成为泌尿外科的一种标准术式之一,它利用远端精索与输精管间的侧支提供血供,而在内环以上水平离断精索血管。可以一期手术,亦可分期手术。
③微血管技术的自体睾丸移植术:应用显微外科技术将腹壁大血管与睾丸血管吻合。早期手术可以保护精原细胞及减少恶变。
(7)腹壁重建术:腹壁重建不仅是美观的需要,对患者心理健康及膀胱、肠道、肺功能均有改善作用。尽管外用腹壁支撑措施如腹带等十分有用,更多的人希望应用重建术。Randolph发现患者两侧及上腹部肌肉大多正常,而耻骨上缺陷最明显,这构成了各种成形术的基础。